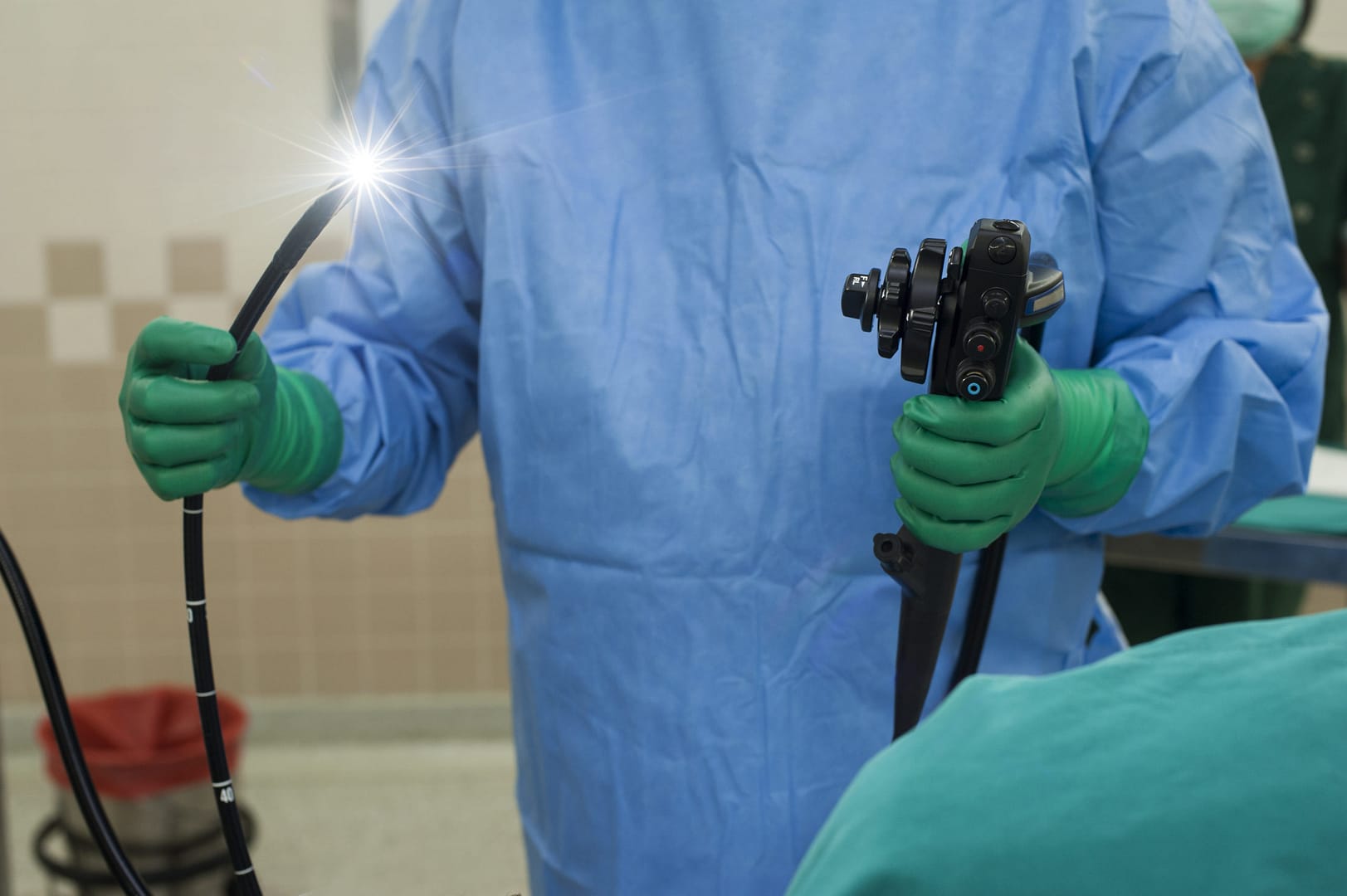
Infections Associated with Reprocessed Duodenoscopes
Duodenoscopes are flexible, lighted tubes that are threaded through the mouth, throat, and stomach into the top of the small intestine (duodenum). They are used during endoscopic retrograde cholangiopancreatography (ERCP), a potentially life-saving procedure to diagnose and treat problems in the pancreas and bile ducts. In the United States, duodenoscopes are used in more than 500,000 ERCP procedures each year.
Duodenoscopes are complex instruments that contain many small working parts. If not thoroughly cleaned and disinfected, tissue or fluid from one patient can remain in a duodenoscope when it is used on a subsequent patient. In rare cases, this can lead to patient-to-patient transmission of infection.
In fall 2013, the Centers for Disease Control and Prevention (CDC) alerted the FDA to a potential association between multi-drug resistant bacteria and duodenoscopes. Upon further investigation, it became clear that these cases of infection were occurring despite confirmation that the users were following proper manufacturer cleaning and disinfection or sterilization instructions.
FDA’s Ongoing Activities
Duodenoscopes are critical to diagnosing and treating severe, often life-threatening diseases. While the overwhelming proportion of procedures with these devices are carried out safely and effectively, the FDA takes the risk of infection very seriously and is working intensively to address it.
Ensuring the safety of reprocessed medical devices for use in multiple patients is a shared responsibility among the FDA and other federal agencies, public health systems, state and local health departments, medical device manufacturers, health care facilities, professional societies, and others. The FDA is actively engaged with many of these stakeholder groups to better understand the causes and risk factors for the transmission of infectious agents and develop solutions to minimize patient exposure.
In October 2015, the FDA ordered each U.S. duodenoscope manufacturer (Olympus, Fujifilm, and Pentax) to conduct postmarket surveillance studies (“522 study”) to better understand how these devices are reprocessed in real-world settings and their impact on duodenoscope transmitted infections. Postmarket surveillance studies are important tools for collecting useful data about a device that can reveal unforeseen adverse events, the actual rate of anticipated adverse events, or other information necessary to protect public health.
On February 26, 2018, the FDA, Centers for Disease Control and Prevention (CDC), and American Society for Microbiology (ASM), together with other endoscope culturing experts, released voluntary standardized protocols for duodenoscope surveillance sampling and culturing. These protocols are an update to the Interim Duodenoscope Surveillance Protocol released by CDC in March 2015, and address the concerns regarding validation of duodenoscope culturing protocols raised in ASM’s April 2015 Policy Statement on Culturing of Duodenoscopes. For health care facilities that choose to implement duodenoscope surveillance sampling and culturing, these protocols can be used to help monitor the quality of a facility’s endoscope reprocessing procedures. Adequate monitoring may reduce the risk of infection.
On March 9, 2018, the FDA issued Warning Letters to all three manufacturers (Fujifilm Medical Systems USA, Inc, Olympus Medical Systems Corporation, and Pentax of America), who make duodenoscopes sold in the U.S. for failure to provide sufficient data to address the postmarket surveillance studies requirements under Section 522 of the Federal Food, Drug, and Cosmetic Act (the Act). All three manufacturers responded to the warning letters and submitted plans that outlines how study milestones will be achieved including enrolling new sites and collecting samples.
On December 10, 2018, the FDA issued a Safety Communication to provide interim results from the ongoing mandated postmarket surveillance studies of duodenoscopes reprocessing. Interim results from the ongoing postmarket surveillance studies indicate higher than expected contamination rates after duodenoscope reprocessing. Facilities and staff that reprocess duodenoscopes are reminded of the importance of manual cleaning prior to disinfection or sterilization and proper servicing of duodenoscopes.
On April 12, 2019, the FDA issued a Safety Communication to provide an update on the postmarket surveillance study results for duodenoscopes used in Endoscopic Retrograde Cholangiopancreatography procedures (ERCP) since the December 2018 Safety Communication. The FDA is also reminding health care facilities about the importance of strictly adhering to the manufacturer’s reprocessing and maintenance instructions, following best practices, and reporting adverse event information to the FDA.
On August 29, 2019, the FDA issued a Safety Communication to provide an update on the mandated postmarket surveillance study results for duodenoscopes used in ERCP since the April 2019 Safety Communication. This Safety Communication also provides additional recommendations and updates including the FDA:
Recommending that hospitals and endoscopy facilities begin transitioning to duodenoscopes with innovative designs that facilitate or eliminate the need for reprocessing.
Issuing new mandated postmarket surveillance study orders to manufacturers of duodenoscopes with disposable endcaps to gather more information and verify that the new designs reduce the contamination rate. Upon completion of the postmarket surveillance studies, the FDA expects the labeling on duodenoscopes with disposable endcaps to be updated with contamination rate data.
Warning health care facilities that adenosine triphosphate (ATP) test strips should not be used to assess duodenoscope cleaning. To date, the FDA has not evaluated them for effectiveness for assessing duodenoscope reprocessing. Manufacturers of ATP test strips are advised to submit data to support the legal marketing of these strips for this use.
Planning to convene the General Hospital and Personal Use Device Panel of the Medical Device Advisory Committee in late 2019 to further discuss duodenoscope reprocessing.
The FDA continues to actively:
Encourage innovative device designs to make it possible to transition away from fixed endcap duodenoscopes to those with more modern design features that facilitate or eliminate the need for reprocessing.
Learn about the challenges with current reprocessing methods and supports expanding the types of validated methods available to reprocess duodenoscopes.
Evaluate information from multiple sources, including medical device adverse event reports submitted to the FDA, the medical literature, the health care community, professional medical societies, international public health agencies, federal partners and state and local governments.
Communicate recommendations to health care providers and end-users to mitigate the risk associated with infection transmission.
Work with industry as they modify and validate their reprocessing instructions to enhance the safety margin of the methods used to clean, disinfect and sterilize the duodenoscope, specifically all three companies that manufacture duodenoscopes marketed in the US and manufacturers of Automated Endoscope Reprocessors (AERs) marketed in the US that reprocess duodenoscopes as stated in their labeling.
Investigate firms that manufacture duodenoscopes (Olympus, Fuji, Pentax). The FDA issued 483s and Warning Letters describing violations to the Federal Food, Drug, and Cosmetic Act to all three manufacturers and 510(k) status letters to two duodenoscope manufacturers (Fuji and Pentax).
Evaluate the effectiveness of current duodenoscope reprocessing instructions in health care settings.
Collaborate with health care facilities, professional societies, and federal partners to evaluate additional strategies for mitigating infections associated with duodenoscopes.
The FDA will continue to provide additional information to the public as new information becomes available.
Source: FDA.gov